Published on
Anthropic 想让 Claude 当化学家,第一步是教它读核磁
Anthropic 发了一篇博客,说他们正和顶尖化学家一起把 Claude 往实验室里送。第一份白皮书没挑什么花哨的题目,而是选了化学家每天都要做的苦活——读核磁谱图。我读完之后,想把它讲清楚,顺便聊聊我的几点想法。

我平时关注 AI 在各个专业领域的落地,化学一直是个让我有点好奇又有点怀疑的方向——它太硬核了,门槛高、数据乱,喊"AI 革命"喊了很多年。最近 Anthropic 发了一篇博客《Making Claude a Chemist》,说他们开始认真做这件事,第一份成果挑的是化学家最日常的一项工作:读核磁谱图。我读完觉得挺有意思,决定还是写出来,顺便附上我自己的几点思考。
一、先搞懂:化学家每天到底在跟什么较劲
我们外行常以为化学家就是"做实验、调试剂"。但 Anthropic 这篇文章里有个观察我很认同:化学家很大一部分时间,其实在做翻译——这也正是我的发小 Dr.Wei 的主业。
![]() 同一个分子,化学家要在好几套"语言"之间反复横跳——白板上手画的结构、仪器打出来的读数、数据库里用来检索的查询字符串、专利和论文里的术语记号。每一套都编码着同样的化学,但每一套都要求一种不同的"流利度"。
同一个分子,化学家要在好几套"语言"之间反复横跳——白板上手画的结构、仪器打出来的读数、数据库里用来检索的查询字符串、专利和论文里的术语记号。每一套都编码着同样的化学,但每一套都要求一种不同的"流利度"。
举个文章里的例子——我觉得很妙:一张咖啡因的结构草图,能让化学家一眼看出它跟腺苷(adenosine,让人犯困的信号分子)长得像,于是推断咖啡因提神的原理是"占了腺苷的座位"。可同样这张草图,却没法帮化学家把咖啡因和另外几个长得几乎一样的分子区分开。
而"到底是哪个分子"这件事,差一点就是天壤之别:
- 把葡萄糖里的几根键重新接一下,它就成了果糖——分子式一模一样,进了身体却走完全不同的代谢通路。
- 把一个分子翻成它的镜像,镇静剂可能就变成致畸剂——当年沙利度胺(thalidomide)那场殃及上万名婴儿的灾难就是这么来的。
所以"认准分子"是化学的地基。麻烦在于规模:CAS(全球最大的化学物质登记库)已经收录了超过 2.9 亿 种公开物质,每天还在新增大约 15,000 种。靠人脑和人手在这些表示之间来回翻译、核对、检索,根本追不上。
二、为什么 AI 在化学里"喊了很多年狼来了"
 读到这里你可能会说:这不正是 AI 该干的活吗?没错,这也是我一直纳闷的地方。机器学习被吹成化学的颠覆者,吹了好些年——逆合成分析(从目标分子倒推该用什么原料一步步搭出来)、反应预测、性质估算,工具一直有,可真正天天用的人不多。
读到这里你可能会说:这不正是 AI 该干的活吗?没错,这也是我一直纳闷的地方。机器学习被吹成化学的颠覆者,吹了好些年——逆合成分析(从目标分子倒推该用什么原料一步步搭出来)、反应预测、性质估算,工具一直有,可真正天天用的人不多。
Anthropic 给的判断我很认同:卡点不在算法,在数据。 化学数据有几个老毛病——失败实验(阴性结果)几乎没人记录、格式五花八门、还大量锁在订阅期刊的付费墙和乱糟糟的补充材料里。逆合成就是典型:能用的 AI 工具存在好多年了,可普通实验室、小课题组的化学家平均下来还是不碰它。
那为什么 Anthropic 觉得"现在"不一样了?因为前沿模型变了两点:
- 会看图(多模态)——能直接从论文配图甚至手绘草图里把结构读出来,不必先把分子喂进一个整理好的数据库。
- 会摊开推理——能读懂方法部分、补充材料里"原样发表"的实验细节,而且会把推理一步步写出来,让化学家能审计它怎么想的。
我特别想强调最后这点。在化学这种容错率极低的领域,一个能把推理过程摊给你看、让你逐步核验的 AI,和一个只丢给你一个答案的黑箱,价值完全不是一个量级。Anthropic 自己把话说得很克制:他们不是宣称 Claude 要取代化学家,而是说 Claude 开始能在翻译、回忆、整合这些辅助性工作上搭把手——是对专业判断的补充,不是替代。这种克制,反而让我更愿意认真看下去。
三、他们干的第一件事:拿核磁谱图开刀

白皮书全文 PDF 在 这里。
先科普一下背景。几乎每一个小分子——药、农药、染料、香料、塑料、DNA 片段——能"存在",前提都是有化学家测定过它的结构。可分子小到光学显微镜都看不见,怎么办?只能靠谱学:拿光、无线电波或磁场去"探"它,看它怎么吸收、发射或偏转能量,得到一张图谱,再从图谱反推结构。
 核磁共振(NMR)就是其中最经典的一招,也是合成化学里最磨人的一步:每做出一个化合物,化学家都得手工把谱图上的每一个峰,对应到结构里的每一个原子上。一个个对,对一下午是常事。
核磁共振(NMR)就是其中最经典的一招,也是合成化学里最磨人的一步:每做出一个化合物,化学家都得手工把谱图上的每一个峰,对应到结构里的每一个原子上。一个个对,对一下午是常事。
Anthropic 这份白皮书要回答的问题很直接:这件苦活,Claude 干得过化学家现在用的专业软件吗? 他们设计了一场相当较真的对比测试:
- 三个 Claude 模型:Opus 4.7、Opus 4.6、Sonnet 4.6
- 两个传统软件:ChemDraw、MestReNova(化学家天天用的那种)
- 20 个化合物,全部取自模型训练截止日期之后才发表的预印本(ChemRxiv)——这一步很关键,等于保证模型没在训练时"背过答案"。每篇论文只取第一个被完整表征的新分子,避免挑肥拣瘦。
- 这 20 个分成 4 个结构家族、每族 5 个,每一族专门对应一类不同的核磁难点。
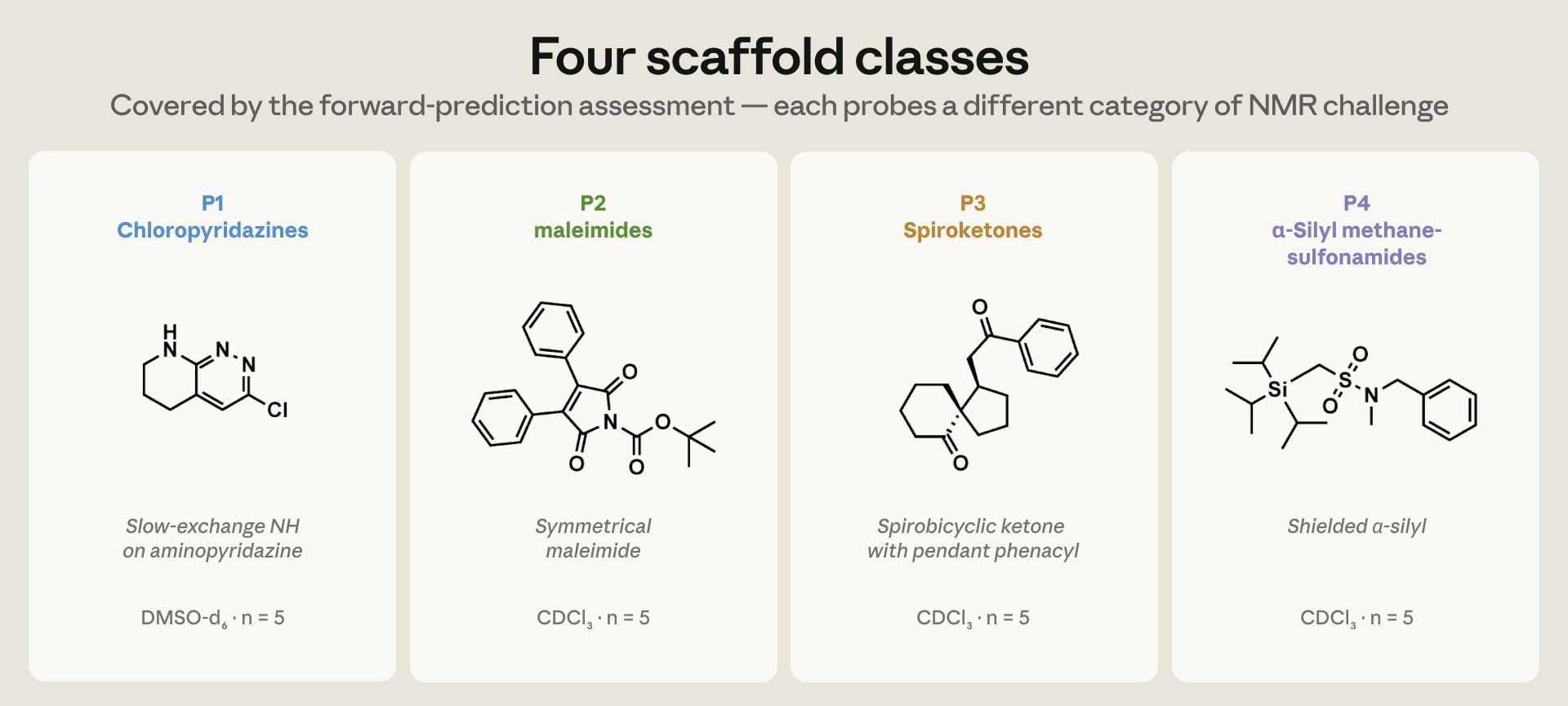
图 1(来自原文):评测覆盖的四类骨架,每一类都踩一种不同的核磁"坑"。P1 氯哒嗪在 DMSO-d₆ 里带个慢交换的 NH;P2 的 Boc-N-芳基马来酰亚胺和 N-Boc 炔酰胺,专考罕见的炔酰胺 α/β 碳;P3 螺酮含非对映异位的 CH₂;P4 α-硅基甲磺酰胺含被屏蔽的硅-α 碳。每族 5 个,共 20 个。
测试的输入是分子的 SMILES 字符串(化学家把分子敲进软件用的那行文本记号),任务是预测一维核磁谱里每个氢、每个碳的峰落在横轴(化学位移,单位 ppm)的什么位置。细节上他们也很讲究:因为样品要溶在溶剂里,而不同溶剂会让峰位轻微挪动,所以每个工具都被要求按论文里实际用的那种溶剂来预测。
四、正着做:通用模型,居然追平了专业软件
先说"正向预测"——给一个画好的结构,模拟它会出什么谱。这是 ChemDraw、MestReNova 的主业。这里有个公平性的小处理我觉得值得一提:语言模型每次输出会有波动,所以每个 Claude 对每个化合物查三次取平均;传统软件每次结果都一样,只跑一次。判定"对"的标准用的是化学家公认的容差——氢 ±0.20 ppm,碳 ±1.0 ppm。
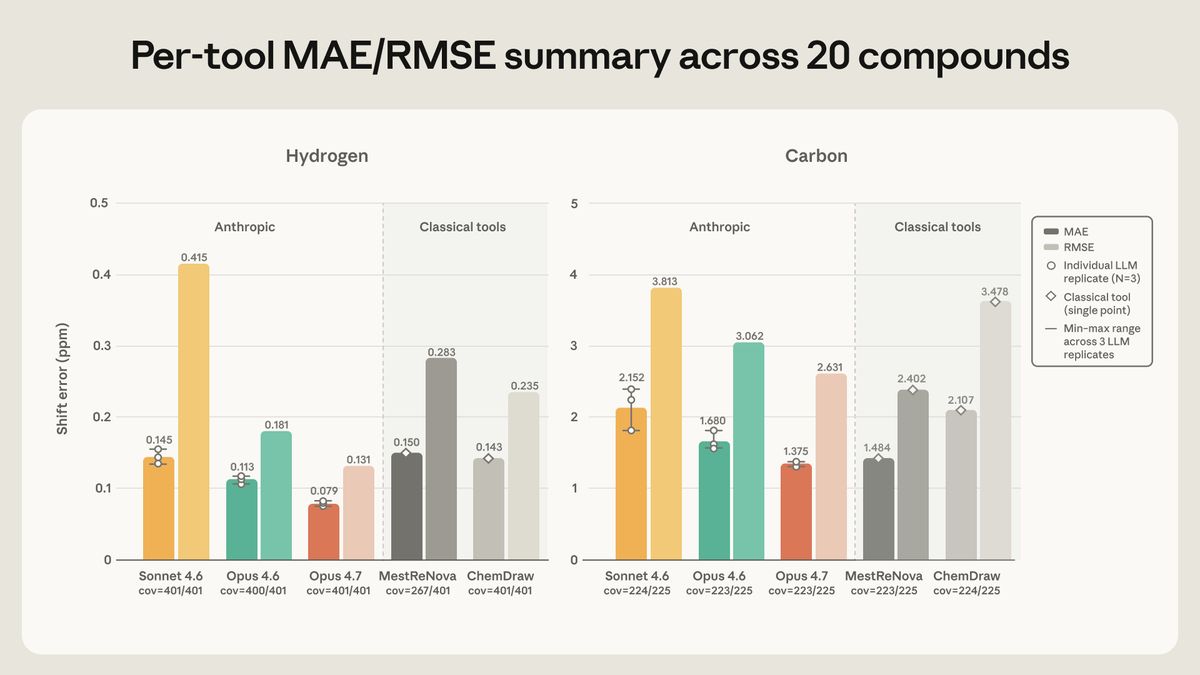
图 2(来自原文):20 个化合物上氢谱(左)、碳谱(右)的逐工具误差。深色是 MAE、浅色是 RMSE。Claude 的柱子带三次重复的范围和散点,传统软件是单点。
结果挺出乎我意料:
- 氢谱:Opus 4.7 最准,平均误差 ±0.079 ppm,连容差窗口的一半都不到,落在窗口内的峰也最多。
- 碳谱:Opus 4.7 和 MestReNova 基本打平(±1.37 对 ±1.48 ppm)。
- 三个 Claude 里,Opus 4.6 居中,Sonnet 4.6 最弱——这个梯度倒是很符合直觉。
最能看出差距的是一个出了名难搞的氢:氯哒嗪家族里的一个 NH 质子,真实位置卡在 6.8–7.9 ppm 这么一个窄带里。Opus 4.7 把它放得略低,但稳定地偏低;Opus 4.6 的猜测撒得到处都是;Sonnet 4.6 直接甩到了 10–13,差了十万八千里。我觉得这个细节比平均分更有信息量——它说明"强模型"的强,很大程度上是稳。
更让我意外的是峰形和峰间距这块。这俩特征里也藏着化学家会顺手读出来的结构信息。Opus 4.7 匹配上实验裂分模式的次数比谁都多;而且三个 Claude 预测的子峰间距,有大约 80% 的时候能精确到半赫兹以内——传统软件 ChemDraw 和 MestReNova 只有 26% 到 35%。
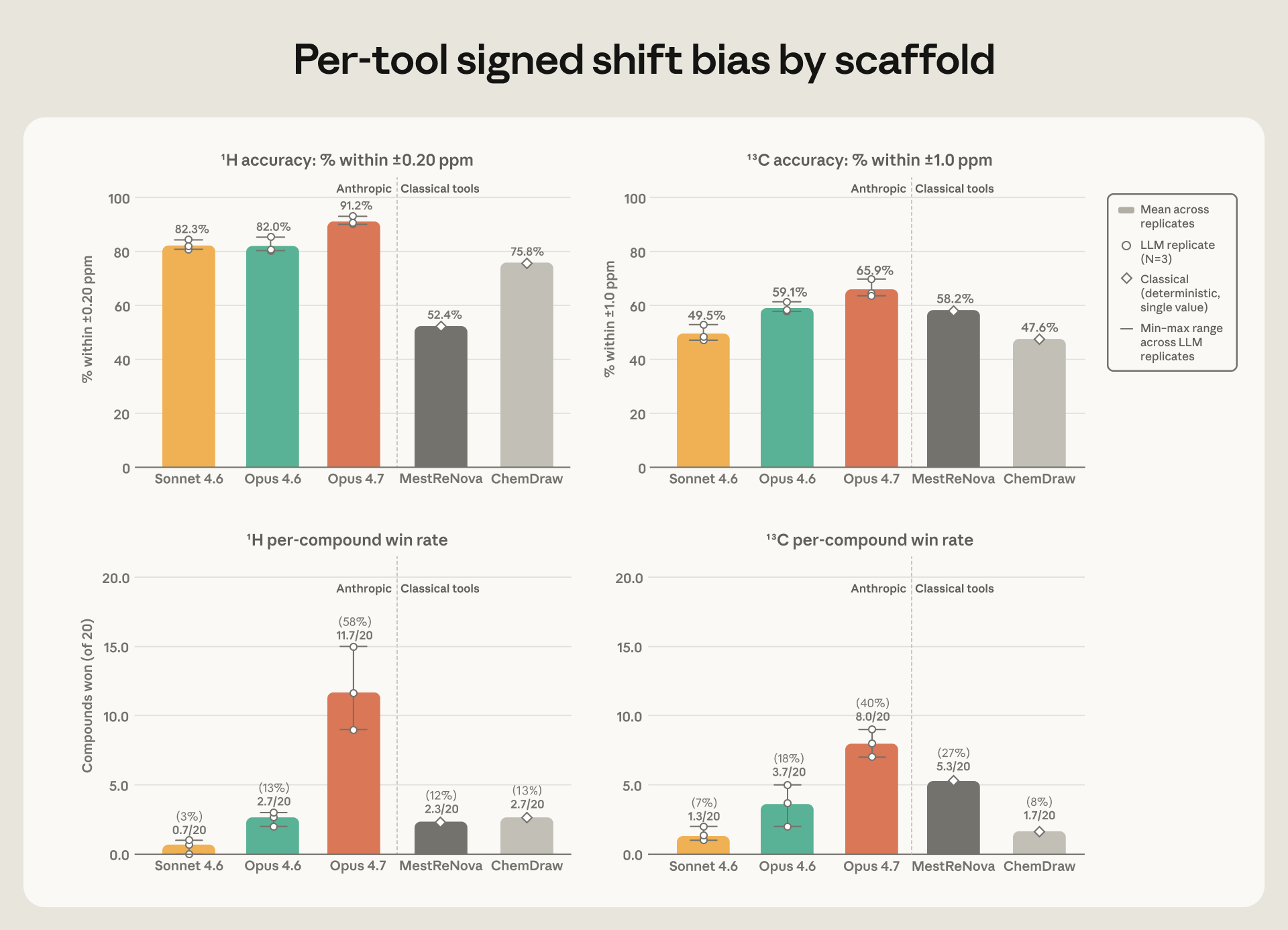
图 3(来自原文):上半是落在容差内的原子占比(左氢、右碳),下半是逐化合物胜率——20 个化合物里,每个工具拿到"最低误差"的有几个。
还有一点我很看重:Opus 4.7 是三次重复里最稳的,它每次跑出来的误差波动,比它和第二名之间的差距还小。换句话说,它赢不是赢在运气好抽中了一次。
五、反着做:从谱图猜结构,这才是真本事

正着做能追平专业软件已经够让我意外了,但接下来这步才是我读这篇文章时眼睛一亮的地方。
正向预测好歹是"已知结构、求谱图"。Anthropic 还试了反过来——从一张实验谱图,反推背后的结构是什么。 这是更难的方向,也是现有软件默认甩给化学家自己做的部分。
他们给 Opus 4.7 出了 15 道结构解析题,每道做三次,要它给最多三个排序后的候选结构。每道提供精确分子式(来自高分辨质谱)加上氢谱、碳谱。15 道按难度分两档:
- 8 道较简单(单环或两个片段拼起来的):只给分子式和谱图,啥提示都没有。
- 7 道更密集(稠环、螺环这类):额外给一个提示——投进反应的起始原料长什么样。
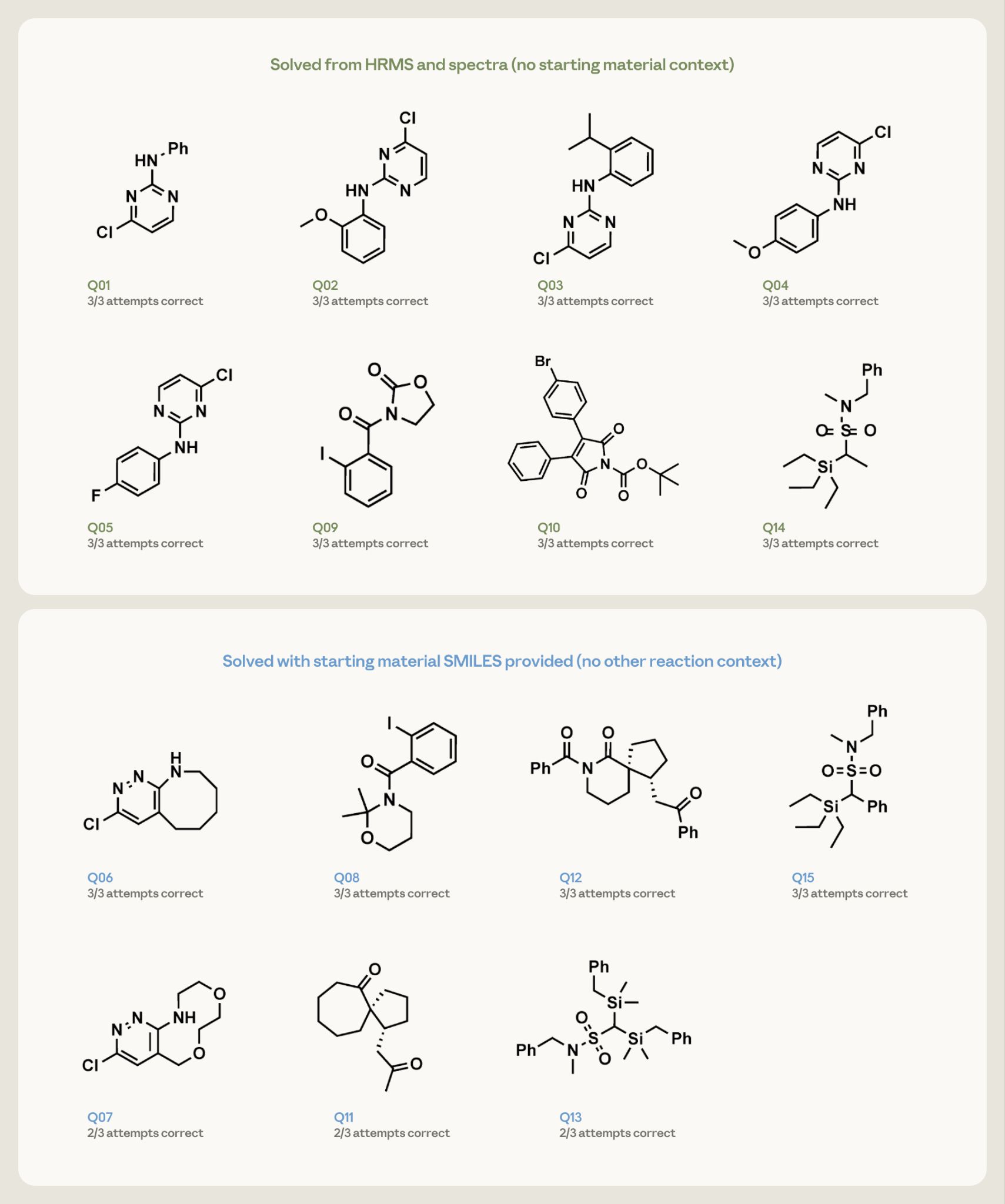
图 4(来自原文):15 道反向题的成绩单。每格是已发表的目标分子加上"3 次里对了几次"。绿框 = 只给谱图和质谱;蓝框 = 多给了起始原料的 SMILES。
成绩单挺漂亮:
- 8 道简单题:只凭谱图和分子式,Opus 4.7 每一道、每一次都答对了。
- 7 道难题(带起始原料提示):其中 4 道三次全对,剩下的也是三次里对两次。
我想强调这件事的分量。专门做结构解析的软件存在几十年了,可它们通常得喂二维核磁(两条轴、出来是等高线图,不是一行峰),还要专门培训、买授权。而 Claude 用的是化学家随手粘进对话框的那种一维峰列表加质谱数据,零配置、零门槛。这个差别,对一个没钱买昂贵软件、也没空学复杂工具的小实验室来说,可能就是"用不用得起 AI"的分界线。
一句话概括这份白皮书:在常规核磁预测上,一个没做过任何化学专门微调的通用模型 Opus 4.7,平均水平已经追平甚至略胜专业软件;而且它还能把题目反着做。
六、泼盆冷水:边界在哪,Anthropic 自己说得很清楚
读到这我其实警觉了一下——这种"通用模型吊打专业工具"的结论,太容易被人拿去过度营销。难得的是,Anthropic 自己把局限摆得明明白白,这点我给个好评:
- 样本太小:正向 20 个、反向 15 个,每类骨架只贡献一种失败模式。结论只能算"有指示性",不是定论。
- 最难的反向题离不开拐杖:最密集的那几个,如果不给起始原料,模型会在推理里反复打转、迟迟不肯收口——所以那 7 道才要带提示。
- 很多骨架压根没测:比如慢交换 NH 杂芳烃只采了氯哒嗪一类,羟基吡啶、氨基噻唑这些近亲都没碰。
- 二维实验和立体化学被有意排除:COSY、HSQC、HMBC 和构型问题不在范围里,因为一维核磁本身就定不了构型——所以复杂天然产物没评。
- 溶剂只测了三种:DMSO-d₆、CDCl₃、D₂O,甲醇、苯、丙酮这些常用氘代溶剂都没覆盖。
他们自己也说,真正过硬的评测应该是几百个化合物、20–30 类骨架、每类至少 15 个,才能把"同类内部的波动"和"工具之间的真实差距"分开。换句话说,这是个有说服力的开头,但远不是终点。 我挺欣赏这种"先把丑话说在前面"的态度,比那些只报喜的 demo 可信多了。
七、他们接下来想啃的硬骨头
Anthropic 在文末列了几个之后想攻的方向,基本就是化学家日常最拖时间的几块:
- 读结构、画结构:把配图、专利、幻灯片、草图里的结构转成机器能读的形式,并在结构和文献里的系统命名之间互转。
- 反应与合成推理:提路线、评路线、挑路线的毛病,预判产物、选择性、副产物。
- 机理:用化学家真用的那套语言——电子箭头、中间体、过渡态——去讲清楚、并检验反应是怎么发生的。
- 读懂化学文献:同一个分子可能被画、被命名、被缩写、被一个编号引用,要能从方法、补充材料、专利里把真正重要的化学抠出来。
他们也坦白,这几件事成熟度差得远:谱学分析已经成熟到能做基准测试了,逆合成规划这种还在"摸边界"。这种"我知道自己还差在哪"的清醒,恰恰是我愿意持续关注这条线的原因。
我的几点思考
读完这篇,我想留下三点自己的想法:
第一,真正的突破不在"更准",而在"换了条路"。 这件事最让我在意的,不是 Opus 4.7 比 ChemDraw 准了多少个 ppm,而是它没靠化学专门微调,纯靠"多模态 + 摊开推理"就绕过了困扰化学界多年的数据难题。数据问题没消失,但"在数据不够的前提下,哪些问题变得可解"这条边界,被往前推了一截。这套思路其实不只对化学有效——任何一个"专业知识散落在图、表、非结构化文本里"的领域,可能都适用。
第二,"能审计"比"能答对"更重要。 化学是容错率极低的领域,一个错峰、一个错构型,代价可能是几周白干甚至安全事故。一个肯把推理一步步摊开、让专家逐步核验的 AI,才配进实验室。我越来越觉得,专业领域的 AI 竞争,最后拼的不是答案多漂亮,而是人能不能信、敢不敢用。
第三,受益最大的也许是"小实验室"。 那些昂贵的专业软件、复杂的二维核磁、专门的培训,本质上是一道资源门槛。如果一个化学家把峰列表粘进对话框就能拿到不输专业工具的结果,那这道门槛就被悄悄抹平了。技术真正的平权,往往就发生在这种"不起眼的日常苦活被接管"的时刻。
当然,这只是第一份白皮书,样本小、边界窄,离"Claude 是个靠谱的化学家"还差得远。但方向我看清楚了——从一张核磁谱图开始,AI 正在一点点学会化学家的母语。这条路还长,我会接着看下去。
参考资料
- Anthropic, Making Claude a Chemist, 2026 年 6 月 5 日:https://www.anthropic.com/research/making-claude-a-chemist
- 白皮书全文(PDF):https://www-cdn.anthropic.com/07441e654ad3dfeb0cd090e9361511562825d012.pdf
- 测试化合物来源的四篇 ChemRxiv 预印本(见原文脚注)
- Anthropic AI for Science 计划:[email protected]